
Large “omics” datasets are increasingly available in public repositories, such as GEO1, ArrayExpress2, Proteome Exchange3 and the GDC4. However, the sheer volume of information requires automated procedures to extract what is biologically relevant for researchers and clinicians. Historically, the NCI60 cell line panel5 (60 cell lines), was the go-to resource that combined phenotypic data with drug response. Later, GDSC6 and CTRP7 have emerged and cover a much greater number of cell lines (987 for GDSC, 860 for CCLE), albeit for a much smaller set of therapies (GDSC1: 320, CTRP: 481) and with a much more incomplete drug-cell line matrix. The next step in the evolution, is the Cancer Dependency Map (https://depmap.org/). The CDM integrates several projects, such as gene expression from CCLE, CRISPR knockouts8, proteomics9 and the PRISM10 drug repositioning screen (578 cell lines, 4686 drugs).
Ironically, when it comes to investigating the sensitivity to drug treatment in a cancer-specific context, limitations of the data become immediately apparent. For example, for the average cancer type only 45 cell lines are available. By contrast, meaningful entities need to be extracted from several thousands of biological features (genes, proteins, mutations) to build a signature. The high dimensionality of the data, coupled with the small sample size makes it very hard to use automated machine learning for feature extraction.
Stefan Naulaerts from the LICR ( Ludwig Institute for Cancer Research, https://www.ludwigcancerresearch.org/ ) institute and Pedro Ballester from the CRCM (Cancer Research Center of Marseille, https://www.crcm-marseille.fr/) are currently working on a workflow that uses the PRISM 19Q3 screen10 and various omics profiles11 to derive small signature sets for each drug and for drug families, using cross-omics approaches and intuitive heuristics to guide the feature selection process. The core of this workflow uses XGBoost12. XGBoost has been previously successfully used for QSAR13.
Their preliminary results suggest that thorough feature selection greatly increased the number of drugs for which a predictive model could be found. In addition, a significant number of the putative signature genes could be retrieved in post-treatment screens as differentially regulated or found in literature as being relevant for the corresponding drug response. In agreement with Corsello et al.10, we confirm that mRNA expression patterns contribute more predictive models than any other data type. Interestingly, proteomics is second, despite having a much smaller set of screened cell lines (and thus having far fewer compounds with 45 or more screened cell lines).
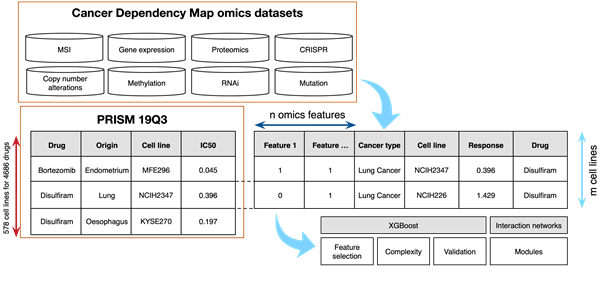
Figure 1: Schematic outline of data and flow. Test set performance for cladribine in lung cancer (bottom left and middle). Cladribine is known to cause cell accumulation at the G1/S phase junction16.
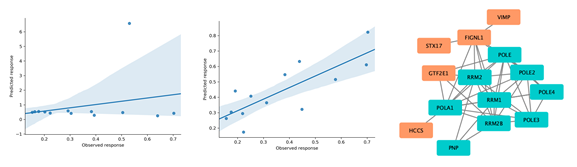
Figure 2: Left: using G1/S-specific transcription pathway signature genes as input. Middle: using guided feature selection for signature generation. Right: protein associations from a public network17 (blue: known targets, orange: selected signature genes)
Key people:
Naulaerts, S. Ludwig Institute for Cancer Research, Brussels, Belgium, de Duve Institute, Université catholique de Louvain, Brussels, Belgium
Ballester, PJ. Cancer Research Center of Marseille CRCM, INSERM, Institut Paoli-Calmettes, Aix-Marseille University, CNRS, F-13009 Marseille, France
References:
1. Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. (2002).
2. Kolesnikov, N. et al. ArrayExpress update-simplifying data submissions. Nucleic Acids Res. (2015). doi:10.1093/nar/gku1057
3. Vizcaíno, J. A. et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature Biotechnology (2014). doi:10.1038/nbt.2839
4. Jensen, M. A., Ferretti, V., Grossman, R. L. & Staudt, L. M. The NCI Genomic Data Commons as an engine for precision medicine. Blood (2017). doi:10.1182/blood-2017-03-735654
5. Shoemaker, R. H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 6, 813–823 (2006).
6. Yang, W. et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, (2013).
7. Seashore-Ludlow, B. et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 5, 1210–1223 (2015).
8. Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. (2017). doi:10.1038/ng.3984
9. Nusinow, D. P. et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell (2020). doi:10.1016/j.cell.2019.12.023
10. Corsello, S. M. et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer (2020). doi:10.1038/s43018-019-0018-6
11. Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature (2019). doi:10.1038/s41586-019-1186-3
12. Chen, T. & Guestrin, C. XGBoost: A Scalable Tree Boosting System. in Proceedings of the 22Nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 785–794 (ACM, 2016). doi:10.1145/2939672.2939785
13. Sheridan, R. P., Wang, W. M., Liaw, A., Ma, J. & Gifford, E. M. Extreme Gradient Boosting as a Method for Quantitative Structure-Activity Relationships. J. Chem. Inf. Model. 56, 2353–2360 (2016).
14. Yang, M., Simm, J., Zakeri, P., Moreau, Y. & Saez-Rodriguez, J. Linking drug target and pathway activation for effective precision therapy using multi-task learning. bioRxiv (2018). doi:10.1101/225573
15. Yang, M. et al. Linking drug target and pathway activation for effective therapy using multi-Task learning. Sci. Rep. (2018). doi:10.1038/s41598-018-25947-y
16. Law, V. et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 42, (2014).
17. Szklarczyk, D. et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. (2015). doi:10.1093/nar/gku1003